
Last August, I wrote about a troubling pattern in inflammation research: brilliant scientists studying arachidonic acid metabolism while remaining “blissfully unaware that half the arachidonic acid story exists in a parallel research universe.” I documented how eicosanoid researchers and endocannabinoid scientists study the same substrate, same concentration ranges, same tissues—yet publish in complete isolation, never acknowledging the other half exists.
I thought maybe, just maybe, that post might spark some introspection in the lipid metabolism community. Perhaps someone would notice they’d been systematically excluding an entire signaling system from their analyses.
Then last month, in January 2026, “The controversial role of linoleic acid in cardiometabolic health” landed in Frontiers in Nutrition. I soon realized…
absolutely nothing has changed.
The same intellectual tunnel vision. The same systematic exclusion of the endocannabinoid system from discussions of omega-6 fatty acid metabolism, despite the ECS being perhaps the most important metabolic pathway driven by dietary linoleic acid.
Let me walk you through what just happened in peer-reviewed nutritional science in 2026, and why it perfectly exemplifies the problem I’ve been documenting on this site.
When a ‘Comprehensive’ Linoleic Acid Figure Forgets the Endocannabinoid System
Berkowitz et al. open their review with Figure 1, presenting what they frame as a comprehensive overview of how dietary omega-6 and omega-3 fatty acids are metabolized into bioactive signaling molecules.
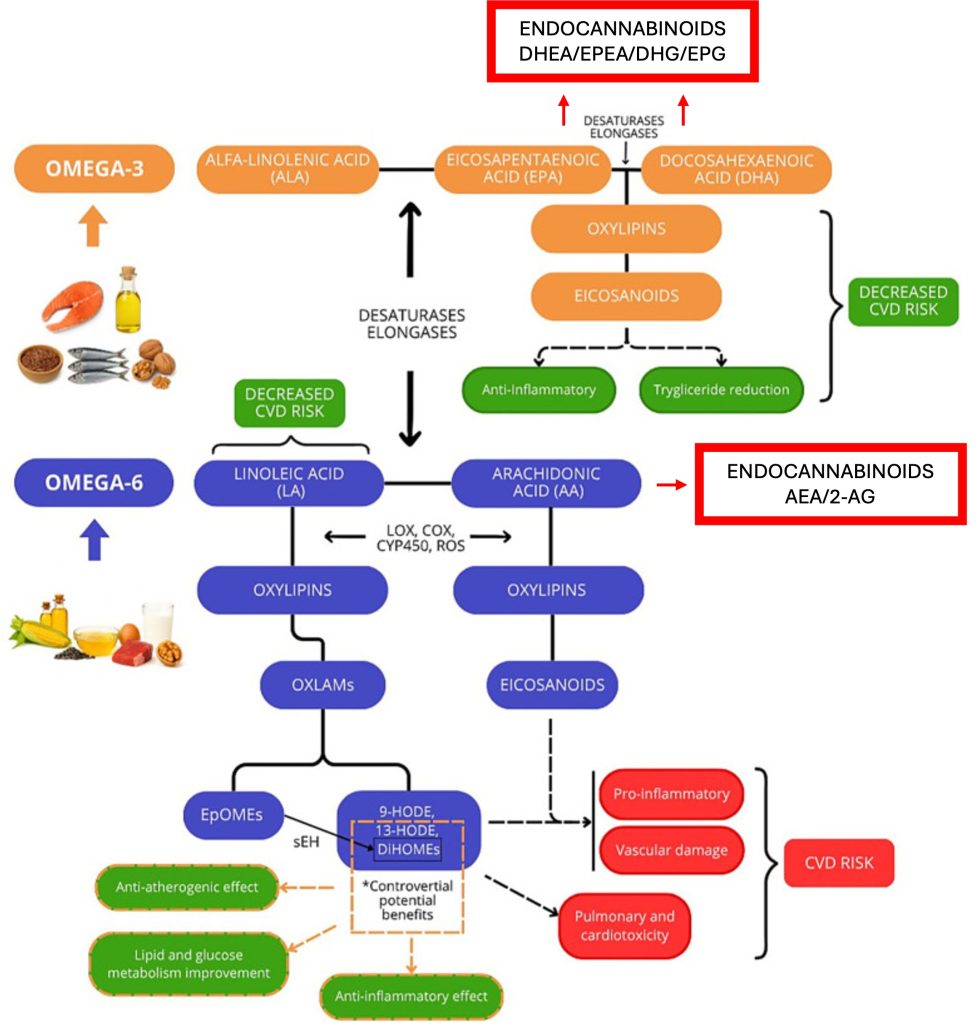
It’s actually quite detailed. They show linoleic acid (LA) converting to arachidonic acid (AA), then carefully trace AA’s transformation into:
- Oxylipins like 9-HODE and 13-HODE
- EpOMEs and DiHOMEs
- Classical eicosanoids via COX and LOX pathways
- “Anti-inflammatory” and “pro-inflammatory” outcomes
The omega-3 side mirrors this: ALA → EPA → DHA → various oxylipins and eicosanoids, with their own color-coded health implications.
Now here’s what I want you to do: scan that figure for the words “endocannabinoid,” “2-AG,” “anandamide,” “CB1,” or “CB2.”
Go ahead. I’ll wait.
Nothing. Endocannabinoids are completely absent from both pathways.
No mention that arachidonic acid is the obligate precursor for anandamide (AEA) and 2-arachidonoylglycerol (2-AG)—the two primary endocannabinoids in your body. No mention that EPA and DHA also generate endocannabinoid-like molecules (DHEA, EPEA) that compete with their omega-6 counterparts for the same biosynthetic machinery. No mention of the CB1 and CB2 receptors that mediate these signals.
Just… nothing.
This would be like publishing a comprehensive diagram of glucose metabolism and forgetting to include insulin receptors. Or mapping cholesterol biochemistry without mentioning LDL receptors.
It’s not an oversight. It’s systematic blindness.
Dietary Linoleic Acid Nearly Triples Liver Endocannabinoids: The Alvheim Studies
Here’s where this goes from “problematic omission” to “borderline scientific malpractice.”
The Berkowitz review cites dozens of studies on linoleic acid metabolism. But Alvheim et al.’s landmark 2012 Obesity paper—showing that increasing dietary LA from 1% to 8% of energy nearly triples hepatic endocannabinoid concentrations—gets exactly one throwaway mention in the context of gut microbiota.
Let me spell out what Alvheim demonstrated, because it’s one of the most mechanistically clear findings in the entire omega-6 literature:
The experimental design: Feed mice identical calories but manipulate LA content (1% vs 8% of energy). Same total fat content in some groups, different LA percentages. Run it for 16 weeks.
What happened to endocannabinoids: Hepatic 2-AG increased 60-80%. Anandamide increased similarly. This wasn’t subtle—it was a dose-dependent explosion of endocannabinoid synthesis.
What happened to the mice: The 8% LA diet (low-fat background) produced similar weight gain and adiposity as medium-fat diets containing 65% more total fat. Plasma leptin doubled. Classic signs of CB1-driven metabolic dysfunction.
The mechanism: Tissue arachidonic acid in membrane phospholipids (AA-PL) increased proportionally with dietary LA. Since 2-AG and anandamide are synthesized directly from membrane-bound AA, more substrate = more endocannabinoid production. Simple biochemistry.
The reversibility: When they added EPA/DHA to the high-LA diet, tissue AA-PL decreased, endocannabinoid levels normalized, and the metabolic dysfunction reversed. Proof of causality.
This has been replicated. Follow-up studies in 2013 and 2014 confirmed the finding. The Fat-1 transgenic mouse—which genetically converts omega-6 to omega-3 fatty acids—shows 50% reduced tissue AA and 86% reduced liver 2-AG despite eating an identical obesogenic diet as wild-type controls.
The evidence couldn’t be clearer: dietary linoleic acid drives endocannabinoid system hyperactivity through tissue arachidonic acid accumulation, and this mechanism directly promotes metabolic disease.
And Berkowitz et al.’s discussion of this? One sentence: “high-LA diets have been shown to induce dysbiosis, increase susceptibility to colitis, and disrupt intestinal endocannabinoid signaling”.
That’s it. Relegated to a microbiome footnote. No discussion of the tripled liver endocannabinoids. No acknowledgment this might be relevant to their stated topic: linoleic acid and cardiometabolic health.
The pattern I documented in my inflammation research post? It’s not just about eicosanoids and endocannabinoids existing in parallel universes. It’s that even when researchers cite endocannabinoid studies, they somehow fail to integrate the implications into their narrative.
The Plasma-Tissue Shell Game
Now, Berkowitz et al. do address one important mechanistic question: doesn’t dietary LA increase tissue arachidonic acid, which would drive all those inflammatory pathways they’re worried about?
Their answer, repeated throughout the review: “intakes above 2% of calories do not significantly increase AA levels, as its production saturates at low LA intake”.
This becomes their get-out-of-jail-free card. See? LA doesn’t increase AA. Therefore LA is safe. Case closed.
As I explained in my UK Biobank analysis, this argument is technically true for plasma AA measured in short-term feeding studies. Delta-6 desaturase saturates around 2-3% LA intake, limiting conversion efficiency. Feed someone 8% LA instead of 2%, and their plasma AA barely budges over a few weeks.
But here’s what they’re not telling you—the sleight of hand that makes this entire argument misleading:
Plasma reflects weeks. Adipose tissue reflects years.
Red blood cell fatty acids integrate ~120 days of exposure. Adipose tissue integrates years. When Baylin et al. measured diet-tissue correlations for LA, they found 0.43 in whole blood, 0.52 in adipose, 0.41 in plasma. These are moderate correlations, meaning tissue composition is not simply mirroring recent dietary intake.
Tissue AA accumulates through multiple pathways, not just LA conversion:
- Direct dietary AA from meat, eggs, poultry (bypasses the desaturase bottleneck entirely)
- Inflammatory PLA2 activation releasing AA from membrane stores
- Chronic metabolic state-dependent partitioning over years
- Obesity-induced upregulation of AA metabolic pathways
Weldon et al. proved this experimentally: increasing dietary LA had minimal effect on mouse tissue AA, but supplementing arachidonic acid directly produced robust tissue accumulation. Same result as human trials.
And tissue AA unequivocally IS elevated in obesity and disease:
- Adipose tissue AA: highest quartile has 20-fold higher odds of abdominal obesity
- Cardiac tissue: “sky-high” AA in obese patients
- Brain: upregulated AA metabolism in Alzheimer’s disease
- Serum AA metabolites: elevated in coronary artery disease, correlating with inflammation
Here’s a critical mechanism: Courville et al. published a 2023 controlled feeding study showing that lowering dietary LA decreased plasma LA but did NOT decrease plasma AA or endocannabinoids in women with overweight/obesity. The tissue pools are already established and self-sustaining.
So when Berkowitz et al. lean on “LA doesn’t increase plasma AA in short-term studies” as evidence that LA is metabolically benign, they’re playing a shell game—showing you one compartment (plasma) over one timescale (weeks) while ignoring the compartment that actually matters (tissue) over the timescale where damage accumulates (years).
Obesity IS Endocannabinoid System Hyperactivity: What the Review Missed
If Berkowitz et al. had done their homework on the endocannabinoid system—or if peer reviewers had noticed the glaring omission—here’s what should have been front and center in their review:
Obesity is definitionally characterized by endocannabinoid system hyperactivity.
This isn’t controversial. It’s textbook:
- Plasma levels of 2-AG and anandamide are elevated in obese humans and correlate with BMI and metabolic markers
- Adipose tissue from obese individuals shows increased CB1 receptor expression
- Visceral fat has higher CB1 density than subcutaneous fat—explaining the particularly pathological nature of belly fat
- Hepatic endocannabinoid levels are elevated, attributed to decreased FAAH (the degradation enzyme)
And this isn’t passive correlation—CB1 activation directly drives metabolic dysfunction:
- Increases fat mass independent of food intake (direct lipogenic signaling)
- Promotes hepatic steatosis even in calorie-controlled conditions
- Impairs insulin signaling and glucose tolerance
- Creates leptin resistance by interfering with hypothalamic leptin signaling
A 2009 review stated it plainly:
“The obese state is characterized by ECS hyperactivity. CB1 activity in the brain facilitates overeating and fat storage, and these signals presumably act synergistically with direct CB actions in multiple other tissues to exacerbate glucose and lipid dynamics”.
de Kloet and Woods wrote in their 2009 Endocrinology review
The Rimonabant Proof Nobody Wants to Talk About
The cleanest proof that ECS hyperactivity causes (not just correlates with) obesity came from the RIO trials—massive phase III studies involving over 6,000 obese subjects treated with rimonabant, a CB1 receptor antagonist.
Results:
- Weight loss: −6.3 kg vs −1.6 kg placebo over 2 years
- Waist circumference: −6.1 cm reduction
- Here’s the critical finding: 50% of the improvements in triglycerides, HDL cholesterol, and insulin sensitivity occurred independent of weight loss
That last point deserves emphasis. Blocking CB1 receptors improved metabolic parameters beyond what the weight loss alone could explain. This proves that hyperactive CB1 signaling has direct metabolic effects separate from its role in energy balance.
Rimonabant was eventually withdrawn for psychiatric side effects (turns out blocking central CB1 makes people depressed and suicidal). But that withdrawal doesn’t invalidate the metabolic findings—rather it validates that the ECS is fundamentally involved in obesity pathophysiology. The field has since moved toward peripheral CB1 antagonists that don’t cross the blood-brain barrier.
So here we have the mother of all smoking guns: pharmacologically blocking the hyperactive endocannabinoid system reverses metabolic disease. And a 2026 review on linoleic acid and cardiometabolic health? Doesn’t mention it. But the ECS-obesity connection goes even deeper—extending from before birth through multiple generations.
Connecting the Dots: mTORC1, Maternal Diets, and the Substrate-Driven Model
Regular readers know I’ve been building a substrate-driven endocannabinoid system framework for understanding how dietary fats control ECS tone. The pieces keep falling into place:
Just last year, we learned that linoleic acid directly activates mTORC1—the master growth regulator—through FABP5 binding. This LA→FABP5→mTORC1 pathway operates independently of insulin signaling, providing a direct nutrient-sensing mechanism. And the ECS and mTORC1 are deeply interconnected: CB1 activation promotes mTORC1 activity in adipocytes and hepatocytes, driving lipogenesis.
Then there’s the transgenerational story. I’ve documented how maternal obesity reshapes breast milk’s endocannabinoid landscape: mothers with obesity and excessive gestational weight gain have significantly higher concentrations of AEA and 2-AG in their breast milk, positively correlated with maternal BMI. But it’s not just endocannabinoid content—maternal body fat itself acts as a “metabolic filter” that reshapes how dietary PUFAs transfer into milk, with higher adiposity mothers showing lower DHA despite similar diets.
The mechanism goes deeper still: maternal obesity disrupts the omega-6/omega-3 balance in breast milk, with overweight/obese mothers showing 50% lower omega-3 and omega-6 PUFAs in plasma but paradoxically higher total PUFAs in milk—yet with sharply reduced DHA in hindmilk and elevated arachidonic acid, skewing the AA-to-DHA ratio toward pro-inflammatory signaling. This creates uneven nutritional exposure: babies get more pro-inflammatory substrate (AA) and less protection from DHA, especially in the nutrient-rich hindmilk that drives growth.
In animal models, maternal high-fat diets rich in omega-6 reprogram offspring endocannabinoid systems, creating sex-specific changes in CB1/CB2 expression and dopaminergic signaling that persist into adolescence—programming offspring for enhanced hedonic consumption and obesity risk.
And this substrate-driven ECS dysregulation isn’t limited to development. As I wrote in August, obesity’s metabolic fingerprint IS endocannabinoid system dysfunction. New metabolomics data from sleep apnea patients revealed that the most distinctive metabolic changes in obesity weren’t respiratory—they were amplified ECS signaling. Excess fat creates a persistent molecular signature: elevated endocannabinoid precursors flood the system, the ECS shifts into overdrive, raising appetite, accelerating fat storage, and priming the body for inflammation.
The infant’s ECS is being programmed during critical windows by:
- Substrate availability (AA, DHA, EPA) in maternal milk—altered by maternal adiposity acting as a metabolic filter
- Endocannabinoid content (AEA, 2-AG) directly transferred through milk—elevated in maternal obesity
- The omega-6/omega-3 ratio driving synthesis capacity—skewed by Western diets high in LA and maternal metabolic dysfunction limiting DHA transfer
These developmental effects may explain transgenerational metabolic programming—why obesity begets obesity even when offspring don’t share the same adult diet. The cycle continues: obese mothers → altered milk composition → infant ECS programming → childhood obesity → adult obesity → altered milk for the next generation.
All of this—LA→mTORC1 activation, maternal ECS programming via milk composition, obesity’s metabolic fingerprint of ECS hyperactivity, tissue substrate accumulation driving adult ECS tone, RBC-AA as an actionable biomarker, Alvheim’s endocannabinoid elevation, Fat-1 genetic proof, rimonabant metabolic reversal—forms a coherent, mechanistically grounded model of how dietary omega-6 fatty acids control metabolic health through the endocannabinoid system across the entire lifespan: from prenatal development through infancy, childhood, adulthood, and into the next generation.
And contemporary lipid reviews? Pretend none of it exists.
Why This Keeps Happening: Institutional Blindness
I’ve thought a lot about why this pattern persists. As I wrote in my inflammation research critique, it’s not stupidity. These are smart researchers publishing in good journals.
It’s systematic institutional structure:
1. Disciplinary silos: Nutrition science trains people in eicosanoid biochemistry. Neuropharmacology trains people in endocannabinoid signaling. They publish in different journals, attend different conferences, cite different literature. The communities never overlap.
2. Grant structure: NIH institutes are organized by organ system and disease category. PUFA research gets funded through NHLBI (cardiology) and NIDDK (metabolism/nutrition). ECS research gets funded through NIDA (addiction) and NIMH (mental health). Proposals spanning both get lost between review panels.
3. Stigma: Despite 30+ years since the CB1 receptor was cloned, “cannabinoid” research still carries stigma. Nutrition researchers avoid the topic to maintain mainstream credibility. I’ve had lipid biochemists tell me privately they find the ECS work fascinating but worry their colleagues will think they’ve gone full stoner science.
4. Complexity avoidance: Acknowledging that LA has context-dependent effects—beneficial when replacing saturated fat in low-substrate individuals, harmful when driving ECS hyperactivity in high-substrate obese individuals—is intellectually uncomfortable. It’s much easier to declare “LA is good” or “LA is bad” and selectively cite supporting evidence.
5. Economic incentives: The vegetable oil industry has strong motivation to fund research emphasizing LA’s benefits (cholesterol lowering) while ignoring potential harms (endocannabinoid-mediated metabolic effects). Industry-funded reviews tend to have remarkable blind spots.
The result? Arachidonic acid doesn’t consult departmental affiliations or journal impact factors before entering metabolic pathways. It simultaneously becomes prostaglandins, leukotrienes, oxylipins, and endocannabinoids. But we study it as if these pathways exist in separate universes.
What Intellectual Honesty Would Look Like
Imagine if Berkowitz et al. had written an intellectually complete review. Their Figure 1 would show:
Omega-6 pathway:
- LA → AA → Endocannabinoids (2-AG, AEA) → CB1/CB2 activation → metabolic effects
- LA → AA → Eicosanoids (via COX, LOX)
- LA → AA → Oxylipins (9-HODE, 13-HODE, etc.)
Omega-3 pathway:
- ALA → EPA → DHA → Omega-3 endocannabinoids (DHEA, EPEA, etc.) → anti-inflammatory, ECS-modulating effects
- ALA → EPA → DHA → Resolvins and other specialized pro-resolving mediators
- ALA → EPA → DHA → Omega-3 oxylipins
Key mechanistic points:
- Omega-6 and omega-3 endocannabinoids compete for biosynthetic enzymes (when one goes up, the other goes down)
- Tissue AA-PL determines endocannabinoid synthesis capacity
- High omega-6/omega-3 ratios drive ECS hyperactivity
- ECS hyperactivity causally contributes to obesity and metabolic syndrome
Clinical implications:
- Plasma LA may be a benign biomarker of vegetable oil intake, but tissue AA accumulation over years drives pathology
- LA recommendations should be context-dependent based on metabolic state and tissue substrate status
- Omega-3 supplementation works partly by diluting tissue AA and normalizing ECS tone
- RBC-AA may serve as an actionable biomarker for ECS-related metabolic risk
This isn’t speculation. This isn’t fringe science. Every claim I just made is supported by peer-reviewed research, much of it from the last five years as the substrate-driven ECS model has matured.
But it requires actually reading the endocannabinoid literature. Which nutritional biochemistry apparently doesn’t do.
The Science Demands Better
Eight months ago, I concluded my inflammation research critique by noting that brilliant scientists studying identical arachidonic acid-driven mechanisms publish in isolation, never acknowledging the parallel pathway exists. I wrote: “This isn’t accidental—it’s systematic, driven by institutional silos and career incentives that reward narrow specialization.”
The Berkowitz et al. review—comprehensive in many respects, published in 2026, peer-reviewed by field experts—proves this pattern not only persists but may be calcifying.
Their Figure 1 systematically excludes endocannabinoids. Alvheim’s landmark findings doesn’t even get a footnote. The rimonabant trials demonstrating CB1-mediated metabolic dysfunction? Never mentioned. The substrate-driven framework connecting dietary PUFAs to ECS tone to obesity? Invisible.
And this is the state of nutritional science in 2026.
I don’t write this just to criticize. I write it because patients deserve better. When someone asks their doctor whether they should avoid omega-6 oils, and the doctor consults reviews like this one, they get an incomplete answer based on an incomplete model.
When someone with obesity and metabolic syndrome tries to understand why dietary changes might help, and the available science ignores the signaling system whose hyperactivity defines their condition, they’re left confused and under-informed.
When researchers design the next clinical trial testing PUFA interventions, and they don’t measure endocannabinoid outcomes because those aren’t “standard metabolic markers,” we lose critical mechanistic insights.
The endocannabinoid system exists. It’s substrate-driven. It responds to dietary linoleic acid within days. Its hyperactivity causally contributes to obesity and metabolic disease. Blocking it reverses metabolic dysfunction.
Any review of linoleic acid and cardiometabolic health that ignores these facts is, by definition, incomplete—no matter how many pages it fills discussing oxylipins.
The science demands intellectual honesty. The disciplines need to talk to each other. And reviews claiming comprehensive coverage of PUFA metabolism need to actually cover all the major signaling systems those PUFAs feed into.
Until that happens, we’ll keep getting reviews that are exhaustively detailed about one half of the story while remaining blissfully unaware the other half exists.
If you’re interested in the substrate-driven ECS framework, check out my previous posts on the UK Biobank omega-6 paradox, how inflammation research ignores the ECS, linoleic acid’s direct mTORC1 activation, and maternal diet’s effects on infant endocannabinoid exposure. This work integrates lipid biochemistry, endocannabinoid pharmacology, and clinical metabolic science to bridge the disciplinary gaps that reviews like Berkowitz’s perpetuate.
Yes. Studies by Alvheim et al. showed that increasing dietary linoleic acid from 1% to 8% of energy nearly triples liver endocannabinoid concentrations (2-AG and anandamide), directly promoting obesity through CB1 receptor activation.
Disciplinary silos, grant structures, stigma around cannabinoid research, and industry funding create systematic institutional blindness where nutrition scientists study fatty acid metabolism without acknowledging endocannabinoid pathways.
The substrate-driven endocannabinoid system framework demonstrates that dietary fatty acids—particularly omega-6 arachidonic acid—directly determine endocannabinoid synthesis capacity, ECS tone, and metabolic phenotype across the lifespan.
Key references:
Berkowitz L, et al. The controversial role of linoleic acid in cardiometabolic health. Front Nutr. 2026;12:1728865.
Alvheim AR, et al. Dietary linoleic acid elevates endogenous 2-AG and anandamide and induces obesity. Obesity. 2012;20:1984-1994; Brit J Nutr. 2013;110:1882-1891.
Courville AB, et al. Dietary linoleic acid lowering alone does not lower arachidonic acid or endocannabinoids among women with overweight and obesity: A randomized, controlled trial. Lipids. 2023;58(6). doi:10.1002/lipd.12382
Multiple reviews on ECS hyperactivity in obesity: Di Marzo, Pagotto, Engeli, Kunos, et al. 2006-2023.
Rimonabant In Obesity (RIO) trials. JAMA. 2006;295:761-775.