
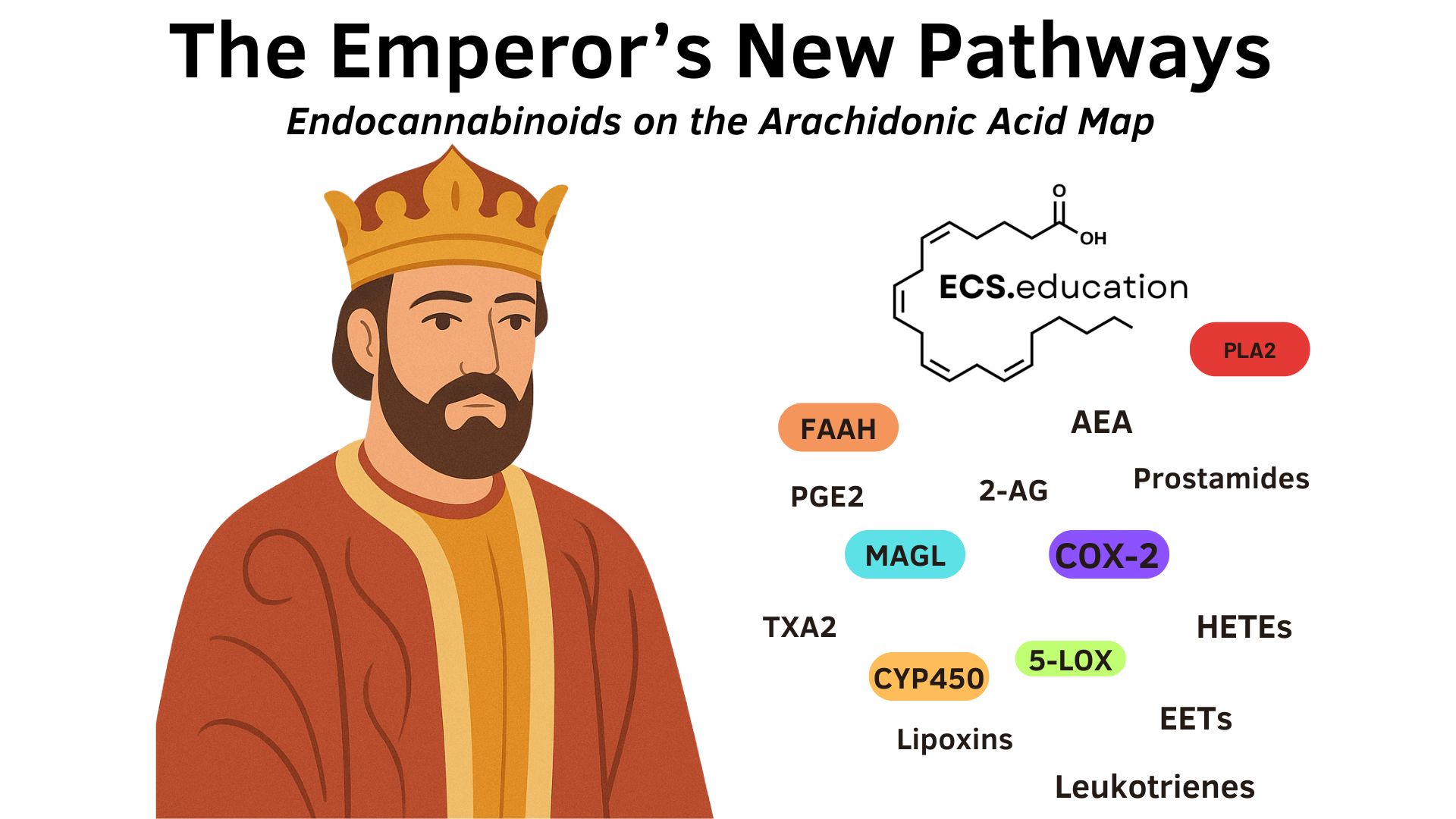
Most NAFLD/MASLD reviews draw a neat AA trident—COX, LOX, CYP—while omitting the endocannabinoid system, even though AEA and 2‑AG are made on demand by NAPE‑PLD and DAGLα/β and rapidly hydrolyzed by FAAH and MAGL back to AA, continuously shuttling substrate between eCBs and eicosanoids in inflamed liver. A new 2025 MASLD review published in expertly details COX‑2/PGE2, LOX‑derived leukotrienes/HETEs, and a CYP2J2‑EET versus CYP4A‑20‑HETE split, but it does not discuss AEA/2‑AG synthesis, FAAH/MAGL recycling, or COX‑2 oxygenation of eCBs into prostamides and PG‑glyceryl esters—gaps that sever causal chains on the same substrate map. COX‑2 oxidizes AEA and 2‑AG into prostamides and PG‑glyceryl esters with non‑classical pharmacology, and substrate‑selective COX‑2 inhibitors can block this endocannabinoid oxygenation while largely sparing AA→prostanoid production, clarifying ‘COX‑2 up’ phenotypes without global PG suppression. MAGL is a major AA gatekeeper for COX flux in brain, liver, and lung; inhibiting MAGL elevates 2‑AG while lowering bulk AA and downstream PGE2/PGD2, revealing a two‑for‑one lever an AA‑only diagram cannot show. Integrating these ECS nodes with the review’s CYP data emphasizes preserving EETs via sEH inhibition as anti‑steatotic and anti‑inflammatory, while recognizing CYP4A‑20‑HETE as insulin‑resistance‑promoting. Finally, lipid sensing can retune GPCR coupling in steatosis; oleic acid was recently identified as an endogenous GPR3 ligand, supporting mechanisms by which lipid load biases hepatocellular signaling states alongside CB1.
Warnings against siloed thinking
In August, the argument was simple: inflammation research keeps ignoring the ECS while drawing lipid networks derived from the same membranes and enzymes, creating half‑maps that mislead readers. This new 2025 MASLD review is a case study in that tunnel vision—deep on COX/LOX/CYP, silent on AEA/2‑AG synthesis and recycling, and silent on COX‑2’s oxidation of endocannabinoids into prostamides and PG‑Gs (Ma et al., 2025). The review diligently catalogs COX‑2/PGE2, LOX‑derived leukotrienes/HETEs, and a CYP2J2‑EET versus CYP4A‑20‑HETE split, but omits AEA/2‑AG synthesis, FAAH/MAGL recycling, and COX‑2 oxygenation of eCBs into prostamides and PG‑glyceryl esters—gaps that break causal chains on the same substrate map.
Why this gap matters
Because eCB generation and hydrolysis return AA to the exact COX/LOX/CYP pool being modeled, excluding ECS nodes fragments pathway logic and hides druggable chokepoints such as MAGL and substrate‑selective COX‑2 control.
Missing nodes and edges
What’s missing on the map are three elements: NAPE‑PLD and DAGLα/β synthesis of AEA/2‑AG, FAAH/MAGL recycling of eCBs to AA, and COX‑2 oxygenation of endocannabinoids to prostamides and PG‑glyceryl esters—routes that feed back into COX/LOX/CYP flux. Adding them reveals a shared AA hub where PLA2 and PLC mobilize AA and DAG; NAPE‑PLD and DAGLα/β generate AEA and 2‑AG; and FAAH/MAGL recycle these lipids to AA that then routes to COX, LOX, and CYP outputs, with COX‑2 also producing prostamide and PG‑G families.
The AA Hub: shared substrate and routing

When AA is mobilized, it does not only feed prostanoids and leukotrienes; it also forms AEA and 2‑AG that are promptly recycled to AA, returning substrate to the same COX/LOX/CYP pool the trident celebrates. COX‑2 has two scripts: beyond AA→PGH2 derivatives, COX‑2 oxygenates AEA and 2‑AG into prostamides and PG‑Gs that carry non‑classical prostanoid biology—signals the MASLD review decided to omit, but that strongly shape “COX‑2 up” phenotypes. Preclinical work across brain, liver, and lung show MAGL is a major AA supplier for COX products in brain, liver, and lung; genetic or pharmacologic MAGL blockade elevates 2‑AG while lowering bulk AA and depressing downstream prostaglandins and thromboxanes, delineating a partition where MAGL, not cPLA2, feeds COX in these tissues. In practice, MAGL inhibition raises eCB tone and simultaneously trims AA‑derived PG output—precisely the two‑for‑one lever an AA‑only diagram cannot reveal.
Layering the review’s COX‑2/PGE2, 5‑LOX leukotrienes/HETEs, CYP2J2‑EETs, and CYP4A‑20‑HETE onto this hub shows how eCB recycling re‑supplies the very substrate those branches consume, as seen in the above figure.
State‑dependent CB1 and cAMP
Endocannabinoids do far more in liver than nudging appetite; they flip GPCR wiring under steatotic cues. Recent work identifies oleic acid as an endogenous GPR3 ligand, supporting a lipid‑sensing route by which steatotic hepatocytes bias CB1 coupling toward Gs and elevate cAMP under lipid load, turning cAMP from a brake into a gas pedal for fatty liver disease (Xiong et al., 2024).
Therapeutic levers revealed by an integrated map
This integrated AA–eCB–eicosanoid map points to three complementary levers: block MAGL to cut the 2‑AG→AA supply and lift endocannabinoid tone, use substrate‑selective COX‑2 inhibitors to stop COX‑2 from oxygenating AEA/2‑AG while sparing classic prostanoids, and preserve CYP‑derived EETs by inhibiting sEH to sustain anti‑steatotic, anti‑inflammatory signaling (Hermanson et al., 2014).
MAGL as AA gatekeeper
In brain, liver, and lung, MAGL is a primary source of the AA pool that feeds COX; inhibiting MAGL raises 2‑AG and simultaneously lowers bulk AA and AA‑derived prostanoids (e.g., PGE2/PGD2), demonstrating that MAGL is a substrate gatekeeper rather than only a catabolic enzyme (Nomura et al., 2011; Cao et al.,2013). This dual effect has been shown across tissues and disease models, including liver inflammation and fibrosis protection with pharmacologic or genetic MAGL blockade (Mulvihill and Komura, 2014).
Substrate‑selective COX‑2 to spare prostanoids
COX‑2 not only converts AA to PGH2 but also oxygenates endocannabinoids, generating prostamides (from AEA) and PG‑glyceryl esters (from 2‑AG); substrate‑selective COX‑2 inhibitors block this endocannabinoid oxygenation while largely sparing AA→prostanoid synthesis, thereby augmenting eCB signaling without the prostaglandin suppression liabilities of traditional NSAIDs (Hu et al., 2008). The mechanism and in vivo efficacy of these SSCIs, including R‑profens and IMMA, are established (Hermanson et al., 2013; Hermanson et al., 2014).
Preserve EETs with sEH inhibition
The CYP epoxygenase arm (e.g., CYP2J2/2C) produces EETs that counter steatosis, oxidative/ER stress, and inflammation; sEH rapidly hydrolyzes EETs to less active diols, so sEH inhibition preserves EET tone and improves NAFLD/MASLD features and portal/liver injury in preclinical models, aligning with the map’s emphasis on sustaining the epoxygenase branch (Liu et al., 2012; Ma et al., 2025).
MASLD review: what it gets right and misses
COX‑2/PGE2 rises in metabolic dysfunction-associated steatohepatitis (MASH), leukotrienes/HETEs drive inflammation/lipogenesis, EETs counter steatosis, and 20‑HETE links to insulin resistance. Placing the review’s COX‑2/PGE2, LOX, and CYP branches onto an AA–eCB hub exposes how FAAH/MAGL and COX‑2’s eCB oxygenation govern flux into both classical prostanoids and non‑classical prostamide/PG‑G families.
Closing remarks
Talking about MASLD without the AA–endocannabinoid loop—and without acknowledging CB1’s state‑dependent cAMP switch—creates half‑maps that misdirect biomarkers and drug development; integrate AEA/2‑AG, FAAH/MAGL, prostamides/PG‑Gs, and EET preservation to reveal actionable control points.
References
- Cao Z, Mulvihill MM, Mukhopadhyay P, et al. Monoacylglycerol lipase controls endocannabinoid and eicosanoid signaling and hepatic injury in mice. Gastroenterology. 2013;144(4):808-817.e15. doi:10.1053/j.gastro.2012.12.028
- Hermanson DJ, Hartley ND, Gamble-George J, et al. Substrate-selective COX-2 inhibition decreases anxiety via endocannabinoid activation. Nat Neurosci. 2013;16(9):1291-1298. doi:10.1038/nn.3480
- Hermanson DJ, Gamble-George JC, Marnett LJ, Patel S. Substrate-selective COX-2 inhibition as a novel strategy for therapeutic endocannabinoid augmentation. Trends Pharmacol Sci. 2014;35(7):358-367. doi:10.1016/j.tips.2014.04.006
- Hu SS, Bradshaw HB, Chen JS, Tan B, Walker JM. Prostaglandin E2 glycerol ester, an endogenous COX-2 metabolite of 2-arachidonoylglycerol, induces hyperalgesia and modulates NFkappaB activity. Br J Pharmacol. 2008;153(7):1538-1549. doi:10.1038/bjp.2008.33
- Liu Y, Dang H, Li D, Pang W, Hammock BD, Zhu Y. Inhibition of soluble epoxide hydrolase attenuates high-fat-diet-induced hepatic steatosis by reduced systemic inflammatory status in mice. PLoS One. 2012;7(6):e39165. doi:10.1371/journal.pone.0039165
- Ma Y, Jiang J, Zhao C, Wei B, Gao J. Arachidonic acid metabolism in metabolic dysfunction-associated steatotic liver disease and liver fibrosis. Hepatol Commun. 2025;9(9):e0802. Published 2025 Aug 29. doi:10.1097/HC9.0000000000000802
- Mulvihill MM, Nomura DK. Therapeutic potential of monoacylglycerol lipase inhibitors. Life Sci. 2013;92(8-9):492-497. doi:10.1016/j.lfs.2012.10.025
- Nomura DK, Morrison BE, Blankman JL, et al. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science. 2011;334(6057):809-813. doi:10.1126/science.1209200
- Xiong Y, Xu Z, Li X, et al. Identification of oleic acid as an endogenous ligand of GPR3. Cell Res. 2024;34(3):232-244. doi:10.1038/s41422-024-00932-5